Last updated October 30, 2013 Download PDF
Catherine A. Ball, Jake K. Byrnes, Daniel Garrigan, Eurie Hong, Keith Noto, Josh Schraiber, Alisa Sedghifar, Shiya Song, Barry Starr, David Turissini, Yong Wang (in alphabetical order)
1. Introduction
The AncestryDNA service offers two primary pieces of information to customers to aid genealogical discovery. The first, known in the population genetics literature as identity-by-descent (IBD) analysis, identifies pairs of customers with long shared genetic segments suggestive of recent common ancestry. Linking a pair of customers with recent common ancestry can allow them to exchange information regarding their family trees.
The second piece of information, which is the subject of this paper, is called genetic ethnicity or genetic ancestry. Here, we provide our customers with an estimate of the ancient historical origins of their DNA. While this information is less relevant for genealogical research relating to the last five to ten generations, it may reveal intriguing clues about the distant history of one's ancestors. Our customers have overwhelmingly expressed interest in receiving the results of this analysis.
AncestryDNA has employed a team of highly trained scientists with backgrounds in population genetics, statistics, machine learning, and computational biology to develop a fast, sophisticated, and accurate method to estimate genetic ethnicity for our customers. In this document, we describe the technology we use to capture a customer's genetic data and the information this technology provides, the reference panel to which we compare each customer sample, the inference method we apply in order to estimate genetic ethnicity, and finally the extensive testing regime we have developed and employed to assess the quality of our estimates. Specifically, this white paper addresses our updated ethnicity estimation process, which includes refinements to the reference panel and a calculation of statistical uncertainty in the ethnicity estimates.
2. Data
Illumina OmniExpress Ascertainment
At AncestryDNA, we genotype samples on the Illumina OmniExpress platform. This platform is designed to assay a majority of the genome while genotyping only 730,525 SNPs. The SNPs on this array are carefully selected to capture the majority of common genetic variation in European and other worldwide populations. Below, we describe the process by which SNPs are chosen for the array.
First, a comprehensive set of data is assembled to determine the SNPs to include on the array. Three sets of populations genotyped by the Human HapMap Consortium, an international effort to catalog human genetic variation (International HapMap Consortium 2005; International HapMap Consortium et al. 2007), are examined separately: 1) the Centre d'Etude du Polymorphisme Humain (CEPH) and Utah Residents with European ancestry (CEU), 2) Han Chinese in Beijing, China and Japanese in Tokyo, Japan (CHB + JPT), and the Yoruba from Idaban, 3) Nigeria in Western Africa (YRI). This dataset reveals millions of candidate SNPs for the array.
From this large pool of candidates, SNPs are chosen based on allele frequency and linkage disequilibrium (LD). First, the OmniExpress platform is designed to capture only common genetic variation - that is, SNPs with allele frequency > 5%. All rare candidate SNPs are removed from consideration. Based on LD, or the correlation between variation at neighboring SNPs, a final set of array SNPs is chosen to optimally ‘tag’ all neighboring variation. While all populations are examined, the majority of the SNPs included on the array are designed to tag variation in European populations. Note that while variation on the X and Y chromosomes is effectively tagged, 706,393 of the 730,525 total SNPs are exclusively on the autosomes.
To determine how well the selected SNPs capture variability in the genome in each test population, the proportion of SNP variation captured by the final set of OmniExpress SNPs at r² > 0.80 is examined. Due to the ascertainment strategy as well as differing population histories and levels of diversity, performance in each population is slightly different (Fig. 2.1). The array currently performs best in European populations (as expected), and captures the least amount of variation in African populations, which are typically more diverse. At AncestryDNA, we are working to account for these differences in our analyses and to find other ways to better capture variation in other populations.
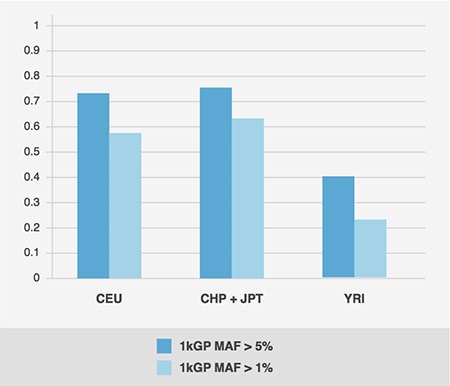
Figure 2.1: Percent Variation Captured at r² > 0.80. Proportion of SNP variation captured at r² > 0.80 in final set of OmniExpress SNPs. SNPs are separated into two classes: common variants (frequency > 5% in the 1000 Genomes project samples) are presented in dark blue, and rare variants (frequency < 1% in the 1000 Genomes project samples) are presented in light blue. As expected, the OmniExpress better tags common variation (Source: Adapted from the following link https://www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/datasheet_omni_whole-genome_beadchips.pdf)
For additional statistics regarding SNPs on the Illumina OmniExpress Platform, see https://www.illumina.com/Documents/products/datasheets/datasheet_human_omni_express.pdf and https://www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/datasheet_omni_whole-genome_beadchips.pdf
Genotyping
Genotyping, the process of using the OmniExpress array to assay a customer's genetic data from DNA that has been purified from a saliva sample, is roughly outlined in Figure 2.2 below. Further details of this process are described at the Illumina website (https://support.illumina.com).

Performance
Both Illumina and AncestryDNA measure a number of statistics to assess the accuracy of the array and genotyping protocol. For studies performed by Illumina, see https://www.illumina.com/Documents/products/datasheets/datasheet_human_omni_express.pdf.
The table below (Table 2.1) shows performance metrics calculated by the scientists at AncestryDNA. First, we measure how often the array returns a genotype for each SNP, or a per-SNP call rate. Taking the average of the per-SNP call rate across 706,393 autosomal SNPs on the array, we find that SNPs successfully return data approximately 99% of the time. To assess the data quality and reproducibility, we assemble 137 pairs of samples which have been genotyped twice on the array. The average per-SNP concordance of genotypes between the first and second runs is over 99.9%.
Though there is a slight difference between data performance estimates produced by Illumina and by AncestryDNA, the results suggest that the vast majority of sites return genotypes and that the genotyping data analyzed in the AncestryDNA service is reproducible.
| Statistic | Value | Description |
|---|---|---|
| Autosomal per SNP Call Rate | 98.7% | The average genotyping rate for each SNP out of the 706,393 we use for analysis. |
| Autosomal per SNP Concordance | > 99.9% | The average rate of agreement between SNP genotypes from 137 pairs of duplicate runs of the same sample. |
SNP Quality Control
We further prune the set of SNPs from the Illumina OmniExpress platform for downstream analyses. First, we remove SNPs from our analysis that have a consistently low call rate. We also remove SNPs with low minor allele frequencies (MAF): SNPs with MAF < 0.02 in each reference population are removed (see next section). We also remove SNPs that do not follow Hardy-Weinberg Equilibrium (see Section 4) with a p-value < 1 x 10-5. Finally, we later remove SNPs in linkage disequilibrium (LD) for our ethnicity estimation procedure (for details, see Section 4).
Sample Quality Control
Finally, at AncestryDNA we perform extensive quality-control (QC) on each customer sample processed, above and beyond the quality control done by our genotyping laboratory. This process, outlined in Figure 2.3 and in the list below, involves checking the percentage of markers successfully genotyped (call rate), comparing the genetically-inferred gender of each sample to the gender reported by the customer during kit activation, confirming that each sample is not a duplicate of a previously processed sample, and quantifying the level of heterozygosity in each sample.
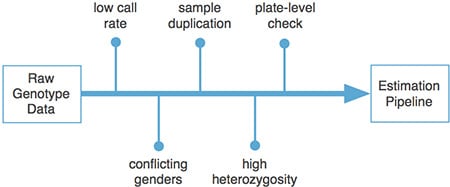
In developing a quality control procedure, we consider the following:
- Samples with low call rates are indicative of failure due to poor sample quality or array failure.
- Samples with conflicting genetic and self-reported gender can indicate misreported gender or sample swapping. Sample swapping can occur at many points along the processing pipeline, including during customer activation. These potential sample-swaps must be manually evaluated, and either recollected or cleared for analysis.
- Although sample duplication is rare, we carefully check each sample to prevent sample duplication.
- High rates of heterozygosity, the percentage of SNPs at which two different alleles are observed for a single sample, can indicate cases in which two samples have been cross-contaminated with one another. Samples with high heterozygosity must be recollected.
Since samples are genotyped in batches of 96, representing a complete genotyping plate, we also carefully consider together all samples from the same plate. There are a small number of potential laboratory errors that can affect an entire plate of samples. Thus, if we observe more than two samples from the same plate with conflicting genders or high heterozygosity, the entire plate of samples will be held for manual examination.
Samples that pass all QC tests will proceed to the analysis pipeline, while those that fail one or more tests will be recollected from customers or manually cleared for analysis.
3. Reference Panel
What is a reference panel and why do we need one?
To determine where your DNA comes from, we need to compare it to a panel of reference samples with known origins. If we can identify samples to which you are genetically similar, and we know the ethnicity of those samples, we can infer your genetic ethnicity from that comparison.
Although there are many approaches for estimating genetic ethnicity, nearly all require a reference panel. The accuracy of our ethnicity estimate is highly dependent on the quality of this reference panel, and thus AncestryDNA has invested a significant amount of effort in creating the best possible reference set of samples.
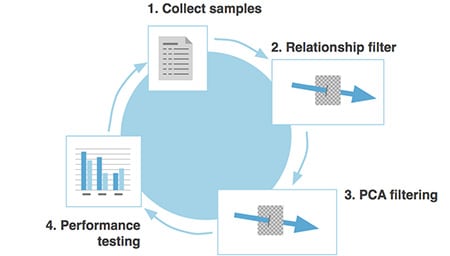
Our current reference panel is version 2 (V2), to distinguish it from the initial Beta release. Version 2 represents a significant step-up in overall quality. Below we describe the steps taken to develop our current V2 reference panel, including sample selection, quality control and testing. The V2 ethnicity update that we describe here is not only an update of the reference panel from the Beta ethnicity version, but also increases the number of global regions representing "source" ethnicity populations from 22 to 26.
Who should be included in the reference panel?
We first create a list of candidate samples to include in the reference panel. Under perfect circumstances, we would construct our reference panel using ancient DNA samples of the true ancestors for each person likely to be an AncestryDNA customer. For example, since many of our customers have ancestors from the United Kingdom, we would prefer to have samples in our reference panel from the Angles and Saxons, who represent the historical populations present in northwestern Europe.
Unfortunately, it is not possible to sample historical populations. We must instead rely on DNA samples collected from individuals alive today who can trace their ancestry to a single geographic location. When asked to trace familial origins, most of us can only reliably trace one to five generations back in time, making it difficult to find individuals with knowledge about distant ancestry. This is because as we go back in time, historical records become sparse, and the number of ancestors we must follow doubles each generation.
Fortunately, knowing where your grandparents are born is often a sufficient proxy for much deeper ancestry. In the recent past, it was much more difficult and thus less common for people to migrate large distances. Because of this, it is frequently the case that the birthplace of your grandparents represents a much more ancient ancestral origin for your DNA.
As a final point on selecting candidates for the reference panel, we prefer to include samples from individuals for whom all ancestral lineages originate in roughly the same location, and thus from the same population. For many individuals, particularly those from the U.S., this is rarely the case. Individuals with recent ancestors from multiple, genetically distinct sources are referred to as admixed. Using samples from admixed individuals in a reference panel complicates analysis. However, this does not mean that a well-designed reference panel cannot be used to identify admixed individuals and assign to them proportions of genetic ethnicity originating in a set of source populations.
AncestryDNA Reference Panel Candidates
In developing the AncestryDNA ethnicity estimation V2 reference panel, we begin with a candidate set of 4,245 samples. First, we examine over 800 samples from 52 worldwide populations from a public project called the Human Genome Diversity Project (HGDP) (Cann et al. 2002; Cavalli-Sforza 2005). Second, we examine samples from a proprietary AncestryDNA reference collection as well as AncestryDNA samples from customers consenting to participate in research. To identify AncestryDNA reference panel candidates from these two sets, family trees are first consulted, and a sample is included in the candidate set if all lineages trace back to the same geographic region. Although this was not possible for HGDP samples, this dataset was explicitly designed to sample a large set of populations representing a global picture of human genetic variation.
Care is taken to include non-admixed, unrelated samples from each participating population, making these samples excellent candidates for our reference panel. In total, our reference panel candidates include over 800 HGDP samples, over 1,500 samples from the proprietary AncestryDNA reference collection, and over 1,800 samples from AncestryDNA customers who have explicitly consented to be included in the reference panel.
Reference Panel Quality Control
We analyze a set of approximately 300,000 SNPs which are shared between the Illumina OmniExpress platform and the Illumina 650K platform used to genotype HGDP samples. After samples with low call rate are removed, we further filter out those who are likely to degrade the performance of the reference panel. Samples are typically removed for one of two reasons.
First, we remove genetically related samples from our panel. When we perform genetic ethnicity estimation, we are interested in computing the probability that an observed allele in a sample's genotype came from each possible source population (see Section 4 below). To do this, we need to estimate the frequency of this allele in each population. Since close relatives contain a significant amount of identical DNA, they do not represent independent samples of alleles in a population. Using related samples can thus distort the estimates of population allele frequencies.
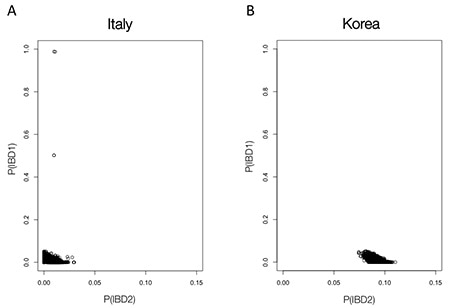
To identify relatives, we use identity-by-descent (IBD) analysis. When two individuals inherit the same allele from a shared common ancestor, the alleles are said to be identical-by-descent. For a pair of samples, we estimate the proportion of genotyped sites for which one allele is IBD (P(IBD1)) using PLINK (Purcell et al. 2007; PLINK). We also estimate the proportion of genotyped sites for which both observed alleles are IBD (P(IBD2)).
For every pair of samples within each population, we examined a scatterplot of computed P(IBD1) and P(IBD2). While the background level of IBD observed within a population depends on the particular history of that population, we use the scatterplots to set population-specific thresholds for P(IBD1) and P(IBD2) (Fig. 3.2). Then, we remove one member of each pair that shows significant sharing.
Second, if the underlying genetic information is not in agreement with the pedigree data for a sample, we remove it. We use Principal Components Analysis (PCA) to identify these outliers. PCA is frequently used for exploratory data analysis in population genetics research (Jackson 2003). Briefly, it is a mathematical method for performing an orthogonal transformation of a data matrix, which in this case is a matrix of genotype data from our candidate samples. The result is a matrix containing a set of linearly orthogonal vectors (principal components) ordered by variance from largest to smallest. When applied correctly to genotype data, following SNP thinning by LD and removal of relatives, the first few principal components capture the genetic variation separating distinct populations (Patterson 2006).
We apply PCA to our global candidate set without relatives, and plot the first two components as a scatterplot. We color each sample by their population of origin, determined by pedigree in the case of AncestryDNA samples and by study sample label in the case of HGDP samples (see Figure 3.3).
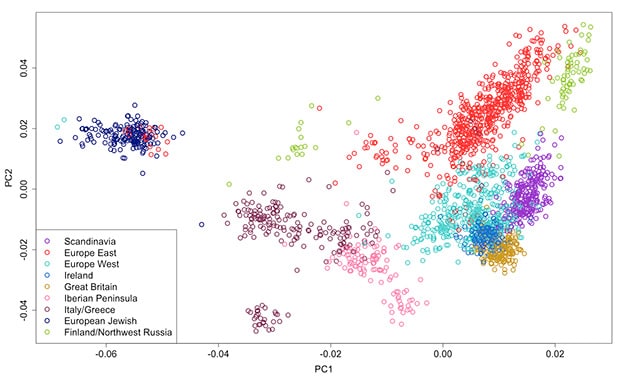
Each population forms a cluster of points, where clusters are separated from one another with respect to genetic distance (and typically also geographic distance). Furthermore, these plots quickly reveal outlying samples who do not appear to be near to other samples from the same population. These are examples where the initial specified population of origin does not agree with the genetic origin represented in PCA space.
We visually inspect candidates for removal based on the first two principal components. Because different collections of samples reveal different amounts of population structure, PCA and outlier removal are repeated for different subsets of data. We first remove outliers at the global level (all samples together), then at the continental level for each continent (e.g. outliers in a PCA using only European samples), then at the regional level for each region (e.g. outliers in a PCA of all Scandinavian samples), and finally at the population level for each population (e.g. outliers from a PCA of Norway).
Iterative Reference Panel Refinement
Finally, we divide our global reference panel into 26 distinct populations, corresponding to distinct genetic clusters based on the PCA and other analyses described above. Each of these populations represents a potential source population with which an AncestryDNA customer may share genetic ethnicity. These regions are described in further detail below.
Before using the reference set to estimate ethnicities of AncestryDNA customers, we perform several experiments to lend support to the quality of this new reference set. This involves testing the performance of our ethnicity estimation procedure on the reference set of samples. (See Section 4 below for details regarding the statistical method used for ethnicity estimation.)
First, we use the new panel to do a leave-one-out analysis. In this experiment, we remove one sample from the reference panel and then use the remaining panel to estimate the ethnicity of the sample that has been removed. We repeat this process for every sample in the panel and then look at the average predicted ethnicity for each region in the set. Figure 3.4 shows the results of this experiment as a box plot.
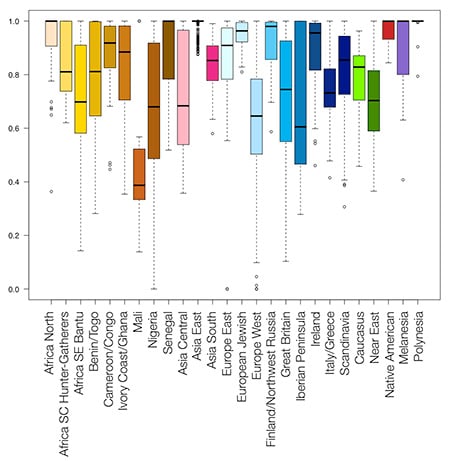
The purpose of this analysis is twofold. First, reference panel samples with poor performance in the leave-one-out analysis were removed. This included samples from individuals whose leave-one-out ethnicity did not represent their ethnic group of origin. (See for instance, Figure 3.5) Second, the leave-one-out plots allow us to define population boundaries and demonstrate our ability to accurately estimate the ethnicities of our reference panel samples using our method (see next section).
V2 Reference Panel
The updated AncestryDNA ethnicity estimation V2 reference panel contains 3,000 samples carefully selected as described to represent 26 distinct global regions (Table 3.1), each with a somewhat distinct genetic profile. As a comparison, our Beta panel represented only 22 distinct global regions.
| Region | # Samples |
|---|---|
| Great Britain | 111 |
| Ireland | 138 |
| Europe East | 432 |
| Iberian Peninsula | 81 |
| European Jewish | 189 |
| Scandinavia | 232 |
| Italy/Greece | 171 |
| Europe West | 166 |
| Finland/Northwest Russia | 59 |
| Africa Southeastern Bantu | 18 |
| Africa North | 26 |
| Africa South-Central Hunter-Gatherers | 35 |
| Benin/Togo | 60 |
| Cameroon/Congo | 115 |
| Ivory Coast/Ghana | 99 |
| Mali | 16 |
| Nigeria | 67 |
| Senegal | 28 |
| Native American | 131 |
| Asia Central | 26 |
| Asia East | 394 |
| Asia South | 161 |
| Melanesia | 28 |
| Polynesia | 18 |
| Caucasus | 58 |
| Near East | 141 |
| Total | 3000 |
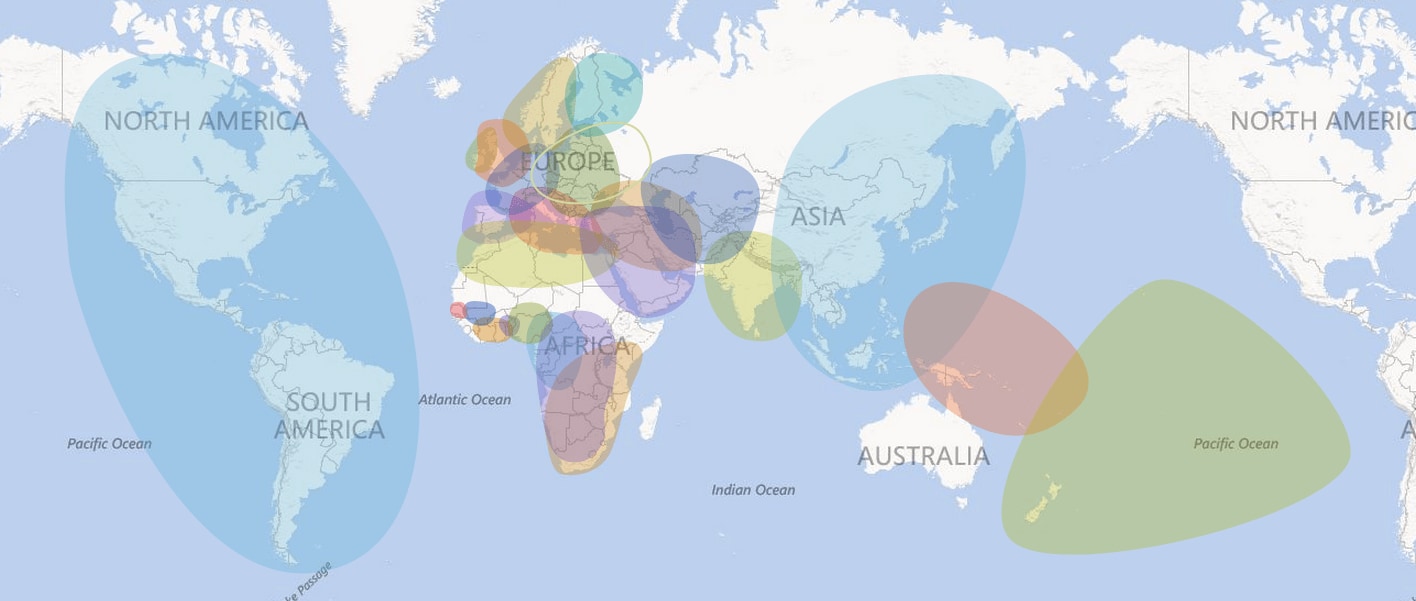
Regional Polygon Construction
As described above, we divide the globe into 26 overlapping geographic regions. Each region represents a population with a somewhat distinct genetic profile. Where possible, we use the known geographic locations of our samples to guide the delineation of regional boundaries. Figure 3.6 shows an example of the information used to define regional polygons.
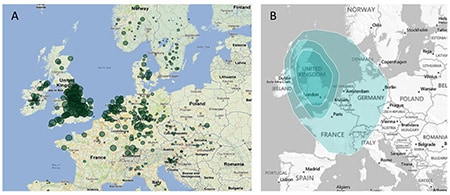
In Figure 3.6A, we show the amount of ethnicity assigned to the Great Britain region for a subset of reference samples with known geographic locations. It is clear from the plot that there is a gradient of ethnicity in this region that is centered in England, tapers off quickly in Ireland to the west, and tapers more slowly into France and Germany to the south and east. Where possible, this information is applied directly to the drawing of regional boundaries (Figure 3.6B) that appear on the maps presented as part of the AncestryDNA product experience.
These polygons appear as nested regions with increasing depth of shading. The more darkly shaded portions represent the more likely physical locations of a customer's ancestors, while the weakly shaded portions represent other possible locations of origin. Each polygon is accompanied by a detailed account of the history of the region.
The map below shows the 2nd tier set of polygons for all 26 ethnic groups, constructed as described above.
We discuss further detailed tests of the performance of the V2 ethnicity panel in Section 5. For methodological details of AncestryDNA's genetic ethnicity estimation, see Section 4.
4. AncestryDNA's Ethnicity Estimation
Introduction
The next step is to estimate a customer's ethnicity based on the DNA of the reference set of samples, as well as the DNA of the customer. We assume that an individual's DNA is a mixture of DNA from a set of "Csource" reference populations. In the example below, a sample gets each allele at each SNP from one of four "Csource" reference populations.

In order to make estimates of genetic ethnicity, we simply use what we know about the frequency of the alleles of those SNPs in the reference populations.
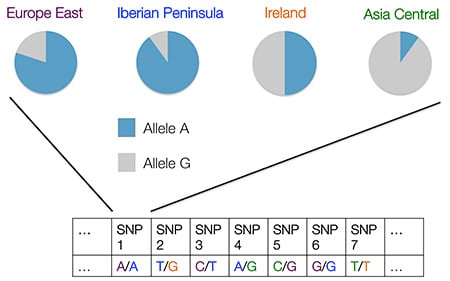
In this example, we are looking at the frequencies of the two alleles at the first SNP in each of the four reference populations. A's are more common in Eastern Europeans and people from the Iberian Peninsula, A's and G's are equally likely in the Irish (from the Ireland region), and G's are more likely in Central Asians. If a sample from an individual has two A's, it seems that Eastern Europe and the Iberian Peninsula are the more likely places from which he or she may have gotten these two alleles. In our example, the individual has gotten 1 A from Eastern Europe, and 1 A from the Iberian Peninsula.
AncestryDNA uses similar reasoning to make our actual estimates, but with a rigorous statistical model that incorporates SNP data from across the genome. The second version of the AncestryDNA ethnicity estimation, in addition to using an updated reference panel, includes greater functionality to estimate statistical confidence of our genetic ethnicity estimates.
Statistical Model
AncestryDNA uses a program called ADMIXTURE, developed by Alexander, Novembre, and Lange (Alexander et al. 2009; https://www.genetics.ucla.edu/software/admixture/). The model estimates the proportions of “membership” in a set of ancestral clusters, or populations, for each sample given his or her genotypes (Prichard 2000). At AncestryDNA, we apply the “supervised” version of the model. Each of the reference populations corresponds to a source population, in which the allele frequencies of each SNP are known and fixed. Both the Beta version of ethnicity estimation, as well as our latest V2 estimates, use the same method.
To introduce the model, we begin by focusing on a single SNP in one sample from an individual in one population. For simplicity, we define the two alleles observed at this SNP as allele R and allele r. Since we will eventually examine more SNPs, samples, and populations, we index a SNP as j, a population as k, and a sample as i.
We first define the probability of observing a particular allele at SNP j in a sample from population k as
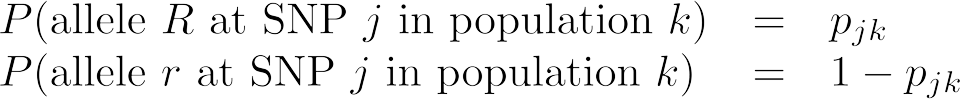
where pjk is the frequency of allele R at SNP j in population k.
These allele frequencies are fixed for each population k based on the allele frequencies in the reference sets, and are easily estimated. The statistical model that we use does not allow for uncertainty in the estimate of allele frequency, and so assumes that the estimate is the true value.
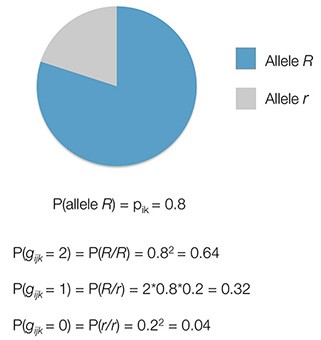
We now introduce the genotype for a sample as gijk: the genotype of sample i at SNP j in population k. For ease, we can define the genotype as the count of R alleles at a SNP position. Thus, gijk can take the values [0,1,2].
Then, the probability of a sample i’s genotype at a SNP j in population k is:

These proportions are based on the assumption of 'Hardy-Weinberg Equilibrium' (HWE), which states that in a population that is randomly mating, the genotype frequencies at bi-allelic sites that are not subject to selection, mutation, or drift will equilibrate at the above stated proportions. In order to perform our estimate, we remove any SNPs that do not follow this pattern of HWE at the 1x10-5 significance level within each homogeneous reference population sample, as mentioned in Section 2.
In the ADMIXTURE model, an individual is from a mixture of K source populations. (Since we examine 26 ancestral reference populations, K = 26 for AncestryDNA’s version 2 ethnicity estimation). Instead of looking only at the probability of a sample’s genotype in one population, we look at all of the possible reference populations given the population allele frequencies.
Below (Fig. 4.4), we explain the intuition for this approach. Take the following concrete example, where we are examining only 2 populations, Europe East and Asia Central. The pie charts show the frequency of allele R in each population.

In this example, if we saw a R/R genotype, the sample seems more likely to have come from Europe East. If we saw a r/r genotype, the sample seems more likely to have come from Asia Central.
Rather than fixing a sample to be from only one source population at a SNP, a sample can actually be a mixture of ethnicities from multiple reference populations. We can give each sample a different “weight” to each ancestral population representing the proportion of their DNA that comes from this ethnicity. In order to allow different populations to have different contributions to a sample’s ancestry, we introduce a new parameter, qik, specifying the proportion of ancestry sample i has from population k. qik is in the range [0,1], and is under the constraint ![]() for all i (since an individuals’ ethnicity is assumed to exclusively come from the set of K source populations).
for all i (since an individuals’ ethnicity is assumed to exclusively come from the set of K source populations).
We can now define the probability of sample i's genotype, conditional only on their ancestry proportions. Below, we have removed the “in population k” description. This is because instead of looking only at one population, we allow a sample’s two alleles to come from multiple source populations.
The equations below are analogous to the single-population equations presented above, except that now the probability of observing each allele is a weighted average of the probability of the allele coming from any possible source population. We sum the allele frequency over all source populations, multiplying by the sample's probability of membership to that ancestral population (qik).
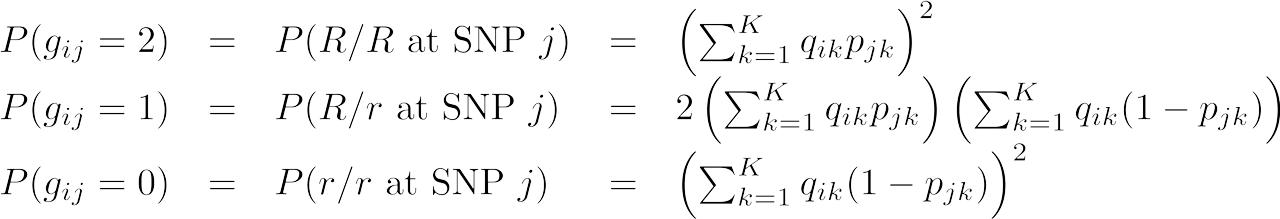
Finally, we can condense the three formulas above into one equation for simplicity, where xij is in the set [0,1,2]:
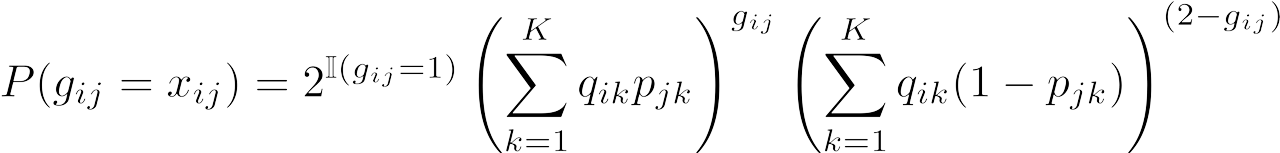
Here, I(gij = 1) is the indicator function, and is equal to 1 when gij = 1 and equal to 0 otherwise.
Take again the concrete example of two ethnicities, Europe East and Asia Central. Again, the pie charts below show the frequency of allele R. Assume for example that the proportion of ancestry from Europe East is 80%, and the proportion from Asia Central is 20% (note that the q’s sum to 1).
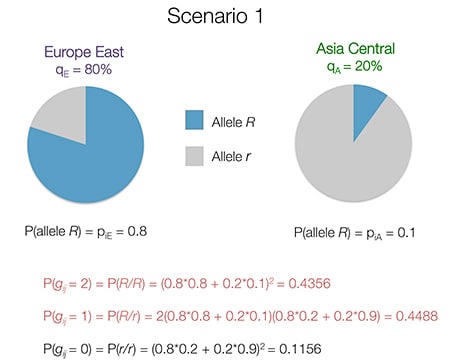
Given these ethnicity proportions, if the genotype of the sample is R/R or R/r, the genotype has a high probability. In contrast, the r/r genotype is far less likely than the R/r genotype.
Below (Fig. 4.6) we switch the sample’s ethnicity proportions to 20% Europe East and 80% Asia Central:
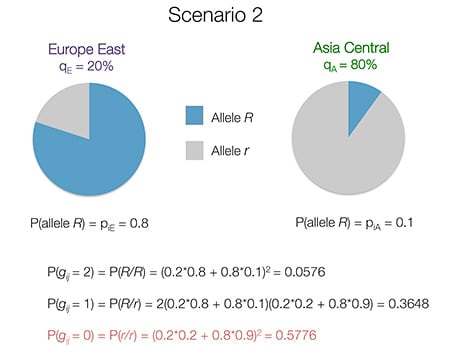
Now, the r/r genotype becomes the most likely, due to the high frequency of allele r in Asia Central. The R/R genotype now becomes less likely.
Clearly, different ethnicity proportions can affect the likelihood of observing a particular genotype. Thus, if we observed a sample with a R/R genotype at this SNP, we might find the first example ethnicity proportions more likely. This is because the R/R genotype is more likely when the ethnicity proportions are 80% Europe East/20% Asia Central than they are when the proportions are 20% Europe East/80% Asia Central.
In reality, however, we don't look at just one SNP position. We look at many highly informative SNPs (see next section), which gives us much more information to learn about a sample's ethnicity. We assume each SNP is an independent observation (see next section), and thus we can multiply the probabilities of the genotype over all SNPs.
We call a sample's genotype at all G SNPs gi (we remove the subscript "j" to indicate that we are looking at all G SNPs. The possible values of a sample's genotype are of the form Xi = [xi1, xi2 …. xiG], where each xij take values in [0, 1, 2]. So, the probability of a sample's full genotype is:
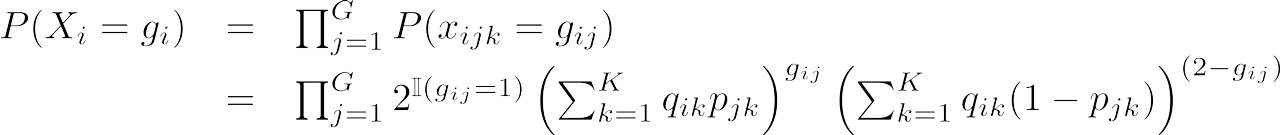
Finally, ADMIXTURE does not just look at each sample separately, but rather at the entire set of samples. Therefore, we can define the full probability of the data (X, which represents the genotypes of all N samples) as
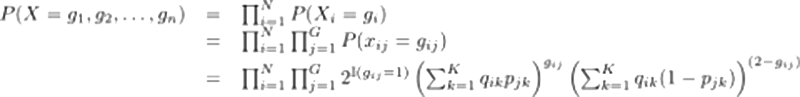
The ethnicity proportions qik can be represented by a matrix Q, which has N rows corresponding to N samples and K columns corresponding to K reference populations. A sample i’s ancestry in population k is the entry Q[i,k] = qik. The allele frequencies can be represented by P, which has G rows (corresponding to the number of SNPs) and K columns; the frequency of SNP j in population k is P[j,k] = pjk.
We estimate Q and P (see below) by maximum likelihood. For the maximization, we are primarily interested in Q. In practice, we maximize the log likelihood:
.png)
We find the qik parameters that make our observed data the most likely to have occurred — i.e., we maximize the likelihood. This involves maximizing over NK parameters for Q, and GK parameters for P.
In the case of 2 populations for sample i, we could search across a grid of values to find the qi1 and qi2 that make the sample’s genotype at all G SNPs the most likely. What we find might look something like this. The x and y axes show the ethnicity proportions, and the coloring shows the “likelihood” of a sample’s genotype.
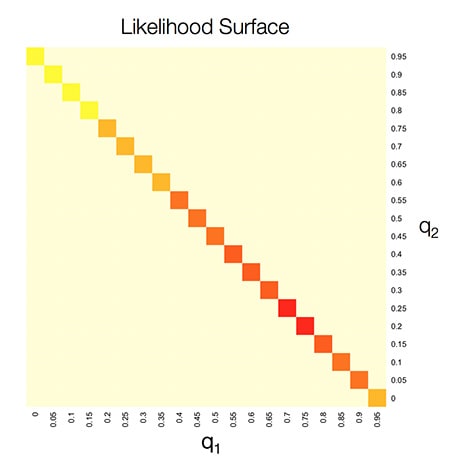
In this example, the sample seems the most likely to have ~70% ethnicity in population 1 and ~30% in population 2. This is similar to our Europe East and Asia Central example. However, instead of just looking at ethnicity proportions of 30% and 70%, we look at all possible combinations of proportions, and over all G SNPs in a sample’s genotype.
In reality, we have to estimate 26N q values for all N samples. Rather than this 2-dimensional plot above, we would need a 26N-dimensional plot. Because it would be inefficient to examine all combinations of the 26N parameters, ADMIXTURE uses an accelerated approach to find the parameters maximizing the likelihood called block-relaxation.
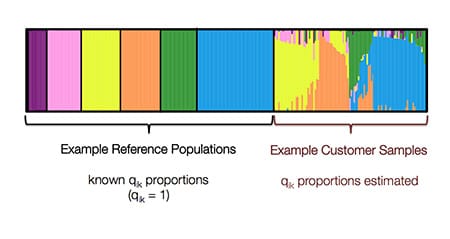
After the entire approach, we obtain estimates of qik for each sample. Below (Fig. 4.8) is an example, where each bar in the bar plot is representative of a sample. Solid bars represent samples included in the reference panel, who are representative of only one ethnicity. Bars with multiple colors represent samples whose ethnicity was estimated using the reference panel and ADMIXTURE, and have membership in multiple ancestral populations.
Assumptions of Admixture Model
There are a number of assumptions of the ADMIXTURE model. First, the model assumes that all SNPs are independent (which makes the multiplication in Equation 4 valid). In reality, SNPs are not actually independent, since we inherit our DNA in chunks, and SNPs can be correlated. (This means that if you have a G at one position, you may be more likely to have an A at the second position instead of a C.)
Since the model requires that SNPs are independent, we remove SNPs that do not appear to be independent using a population-specific window-based LD thinning method using the program PLINK (Purcell et al. 2007) (as mentioned in Section 2). While this means that we are using fewer SNPs in the estimation, we meet the requirements of the model by using an independent set of SNPs. In practice, we use over 100,000 independent SNPs, which effectively captures information from the entire set of over 300,000 SNPs (described in Section 3).
Another assumption that follows from the likelihood equation above (Equation 5) is that all samples are independent, or unrelated. In order to meet this assumption, we preprocess the genetic samples to place any samples from related individuals into separate runs of ADMIXTURE. In a particular run, we also remove any reference samples to whom a customer appears to be related.
It should also be noted that the approach we use is not entirely “supervised,” although we use a supervised version of the algorithm. While the reference populations are set as the “source” populations, genotypes of the tested samples can also influence the allele frequency estimates in the source clusters; i.e., the approach is not fully supervised. This is because the model not only estimates Q, but also P, as a function of both the reference samples and the customer samples (a total of N samples). While ideally the P values should remain stable regardless of the customer samples, the customer samples could slightly change the P estimates from their “true” values.
Customer samples are run in batches of varying sizes; due to the details of the algorithm described above, in theory a customer’s results could vary by batch. Extensive tests have shown that the effect of batch on customer estimates is minimal. This is because the batch size is very small compared to the size of the reference panel. Also, removing related samples from the same batch, as described above, ensures minimal effects on customer ethnicity estimates.
Estimation of Uncertainty
When considering AncestryDNA estimates of genetic ethnicity it is important to remember that our estimates are, in fact, estimates. The estimates are variable and depend on the method applied, the reference panel used, and the other customer samples included during estimation. In AncestryDNA ethnicity estimation version 2, we have added a measure of uncertainty to our ethnicity predictions qik that were not provided in the Beta version.
To do so, we use the bootstrapping calculation that is part of the ADMIXTURE program. The approach has been called bootstrapping in the statistical literature because you do not use statistical tables, simulations, or additional data to get the estimates of variability — you simply use the collected data. Bootstrapping is a statistical technique used to assess the variability of an estimate by repeated estimation of the same quantity using different resampled sets constructed from the available data.
In the case of genetic ethnicity estimates, one way these estimates might vary is if our dataset included a different set of SNPs. We use bootstrapping to estimate the effect of the chosen of SNPs on the uncertainty in our ethnicity estimates. For all samples, we resample “blocks” of SNPs using the default parameters of ADMIXTURE to make a new genome of the same size (called the “moving-block bootstrap”). Blocks, rather than individual SNPs, are sampled to account for any spurious associations between neighboring SNPs.
An important detail is that the blocks of SNPs are sampled with replacement. This means we could sample the same SNP block more than once, or not at all. In the example below, we have not sampled any orange colored blocks, but we have sampled a few coral blocks, and only one blue block.
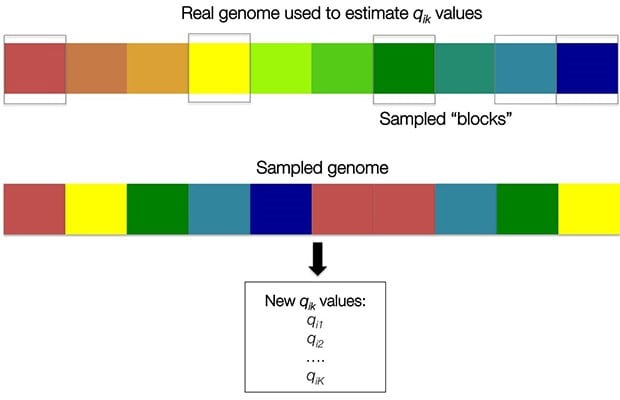
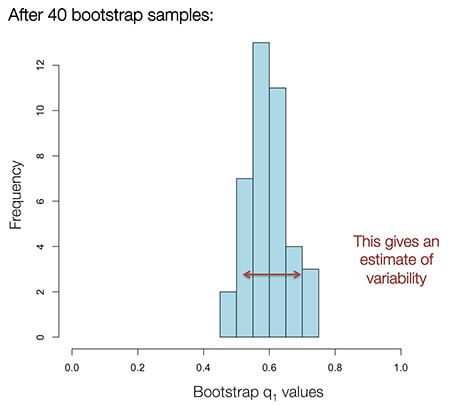
We can then obtain additional Q estimates for each sample based on the SNPs in the new “genome.” After performing bootstrap resampling 40 times, we generate a sample of likely Q values for each sample (see Figure 4.10). For each ethnicity, we report to the customer the mean value of these re-sampled ethnicity proportions (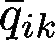 ). Based on the bootstrap samples, we also report a likely range for each estimate. For samples from individuals with no ethnicity in a particular region, reported ranges correctly include 0% greater than 95% of the time.
). Based on the bootstrap samples, we also report a likely range for each estimate. For samples from individuals with no ethnicity in a particular region, reported ranges correctly include 0% greater than 95% of the time.
5. Evaluation
It is critical for us to demonstrate that the AncestryDNA ethnicity estimation V2 reference panel, which estimates ethnicities in 26 global regions, significantly outperforms the Beta reference panel (which estimates ethnicities in 22 global regions) (see Section 3). We perform extensive tests which allow us to evaluate our current performance and confirm that the performance of AncestryDNA ethnicity estimation V2 is much improved. In addition, our analyses guide research for future improvements to AncestryDNA ethnicity estimation.
Comparison to Pedigrees
First, our unique collection of pedigree data allows us to actually measure the similarity between pedigree estimates of ethnicity and genetic estimates of ethnicity. However, pedigrees contain information that is quite different from what we are estimating at AncestryDNA. They show only the locations of a sample’s known ancestors, whereas in genetic ethnicity estimation, we are attempting to estimate the unknown amount of DNA actually inherited from all of a sample’s ancestors. Genetic estimates of ethnicity also go back thousands of years, beyond the end of a pedigree paper trail. Regions identified as “populations” in a pedigree may have been very different thousands of years ago, and so may be represented differently in a genetic ethnicity estimate.
Nevertheless, the agreement between a pedigree and our genetic ethnicity estimate helps us to track improvements to our region boundaries, set of reference samples, and overall algorithms. Therefore, we have assembled several evaluation sets including samples from multiple European regions, samples with DNA from a cross-section of all 26 V2 regions, as well as samples that we believe come from a single one of our 26 regions.
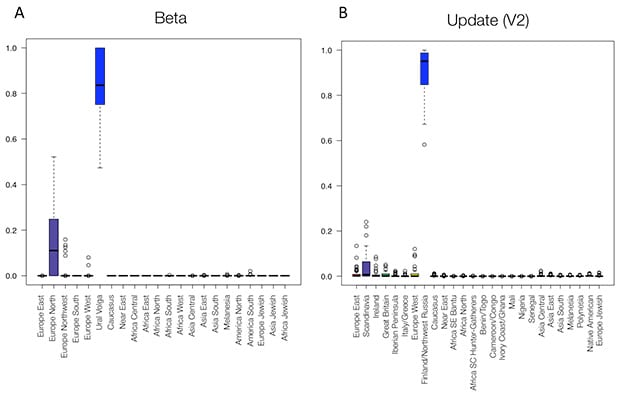
The updated V2 panel outperforms the Beta panel for all of our evaluation sets; see, for example, Figure 5.1. In samples whose known ancestors are only from the Finland/Northwest Russia region (previously referred to as Ural Volga), we more consistently estimate samples to have nearly 100% ethnicity from this region using the V2 panel. The V2 ethnicity update also results in fewer estimates of ethnicity in other regions, such as Scandinavia (previously referred to as Europe North). Other regions show similar patterns in improvement (see also the section below).
Concordance between Relatives
To specifically test the consistency of our ethnicity estimates between relatives, we construct several datasets of samples from related individuals, which should share a predictable amount of genetic ethnicity. For example, because siblings inherit large amounts of the same genetic material from their parents, there is an expectation that their genetic ethnicity estimates should be similar. The same is true for first cousins and parents and children. Since DNA inheritance is a random process, the actual ethnicity percentages for relatives are not expected to be identical, but similar.
Using the new V2 panel, genetic ethnicity estimates show greater overlap of estimated regions between both siblings and first cousins. We also find greater consistency of genetic ethnicity estimates between parents and children when examining the estimates of nuclear families consisting of two parents and one child. With the new V2 panel, fewer than one region on average is present in a child that is not present in a parent.
In conclusion, the ethnicity V2 reference panel improves the consistency of genetic ethnicity estimation for relatives. These tests complement results from the pedigree concordance tests, confirming that genetic ethnicity estimates using AncestryDNA ethnicity estimation V2 are greatly improved from estimates using the Beta version.
Region Assessment
Analyzing samples from individuals whose known ancestors are from only one of our ethnicity regions also allows us to measure how much overlap exists between regions, to track improvements to our region boundaries, as well as to communicate results to our customers.
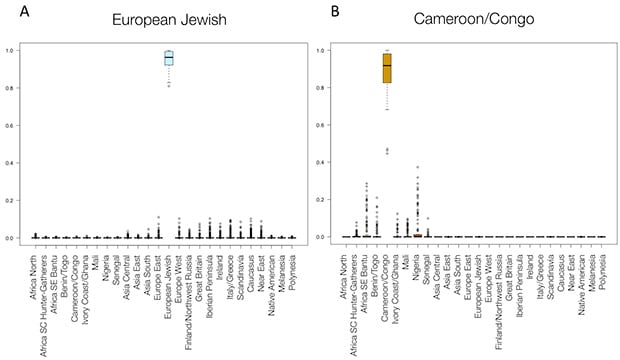
First, we can identify cases where no further development is immediately necessary. For example, AncestryDNA ethnicity estimation V2 accurately identifies genetic ethnicity for the European Jewish and Cameroon/Congo regions, among others (Figure 5.2). In each of these cases, very little ancestry is mis-assigned to other populations, including geographically close neighbors.
We can also use these experiments to identify cases where our current approach could use future improvement. For example, the boxplots in Figure 5.3A show that samples from Great Britain are mis-assigned a significant amount of Europe West ethnicity. Figure 5.3B shows that the reciprocal is also true. Britain and Western Europe are geographically close, and a significant amount of historical migration (and hence, interbreeding) has occurred between these regions. Frequent interbreeding has led to very little genetic differentiation between these two regions, and thus our current approach has less power to identify the true ancestral source for samples from individuals with ancestors from these locations. (We note, however, that estimation for these two regions is improved using AncestryDNA ethnicity estimation V2 as compared to the Beta version.)
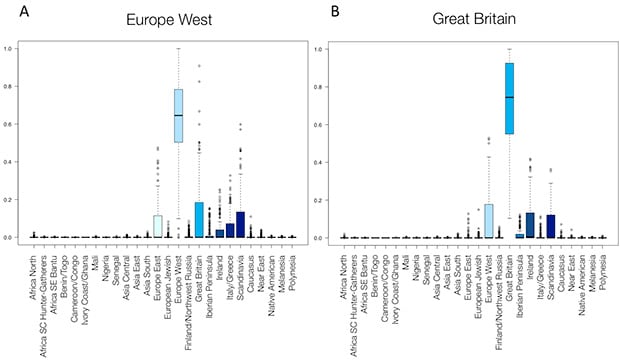
These analyses help us to understand the genetic diversity of the V2 regions and allow us to better communicate these results to our customers (e.g., even if all of your ancestors are British, you can expect to have some amount of genetic ethnicity from adjacent regions). These analyses also aid us in prioritizing future developments for further ethnicity estimation updates.
6. Future Ethnicity Estimation Refinement
While AncestryDNA is extremely proud of the updates in this V2 release of genetic ethnicity estimation, we will continue to improve the product over time. The availability of new data, the development of new methodologies, and the discovery of new information relating to patterns of human genetic variation will all necessitate future improvements to the product.
Each of the steps above represents a critical part of our ethnicity estimation procedure and development. Currently, we are working to further expand our global reference panel for future ethnicity updates. We have already begun genotyping and analyzing samples for a future update which will provide finer-grained estimates of ethnicity. Simultaneously, we are also working to improve our algorithms for ethnicity estimation. Future ethnicity updates will include an improvement to our statistical methodology that will more fully leverage information in genetic data to reveal even more information about population history. Along the way, we always perform thorough testing, involving analyses like those described above. These tests inform the focus of our improvements, and help to refine our improvements as necessary.
Each new release of genetic ethnicity estimation will represent a step forward in our ability to give our customers a complete description of their genetic ancestry and inform them about their ancient genetic origins. We hope that, like the entire team at AncestryDNA, our customers will look forward to these future developments.
7. References
- D.H. Alexander, J. Novembre, and K. Lange. Fast model-based estimation of ancestry in unrelated individuals. Genome Research, 2009. 19:1655-1664.
- Cann HM, de Toma C, Cazes L, Legrand MF, Morel V, Piouffre L, Bodmer J, Bodmer WF, Bonne-Tamir B, Cambon-Thomsen A, Chen Z, Chu J, Carcassi C, Contu L, Du R, Excoffier L, Ferrara GB, Friedlaender JS, Groot H, Gurwitz D, Jenkins T, Herrera RJ, Huang X, Kidd J, Kidd KK, Langaney A, Lin AA, Mehdi SQ, Parham P, Piazza A, Pistillo MP, Qian Y, Shu Q, Xu J, Zhu S, Weber JL, Greely HT, Feldman MW, Thomas G, Dausset J, Cavalli-Sforza LL. A human genome diversity cell line panel. Science, 2002 Apr 12;296(5566):261-2.
- Cavalli-Sforza LL. The Human Genome Diversity Project: past, present and future. Nat Rev Genet. 2005 Apr;6(4):333-40.
- International HapMap Consortium. A haplotype map of the human genome. Nature. 2005 Oct 437(7063): 1299-1320.
- International HapMap Consortium, K. A. Frazer, D. G. Ballinger, D. R. Cox, D. A. Hinds, L. L. Stuve, R. A. Gibbs, J. W. Belmont, A. Boudreau, P. Hardenbol, S. M. Leal, S. Pasternak, D. A. Wheeler, T. D. Willis, F. Yu, H. Yang, C. Zeng, Y. Gao, H. Hu, W. Hu, C. Li, W. Lin, S. Liu, H. Pan, X. Tang, J. Wang, W. Wang, J. Yu, B. Zhang, Q. Zhang, H. Zhao, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature, 2007 Oct 449(7164):851-61.
- Jackson, J.E. A User’s Guide to Principal Components (John Wiley & Sons, New York, 2003).
- Patterson N, Price AL, Reich D. Population Structure and Eigenanalysis. PLoS Genet 2006 2(12): e190.
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000 Jun;155(2):945-59.
- Purcell, S. PLINK v1.07. https://pngu.mgh.harvard.edu/purcell/plink/
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007 Sep;81(3):559-75.